Protein mutations may lead to pathologies by causing protein misfunction or propensity to degradation. For this reason, several studies have been performed over the years to determine the capability of proteins to retain their native conformation under stress condition as well as factors to explain protein stabilization and the mechanisms behind unfolding. In this review, we explore the paradigmatic example of frataxin, an iron binding protein involved in Fe–S cluster biogenesis, and whose impairment causes a neurodegenerative disease called Friedreich’s Ataxia (FRDA). We summarize what is known about most common point mutations identified so far in heterozygous FRDA patients, their effects on frataxin structure and function and the consequences of its binding with partners.
Sunday, February 13, 2022
R. Protein Mutations and Stability, a Link with Disease: The Case Study of Frataxin
Puglisi, Biomedicines 2022, 10, 425. doi:10.3390/biomedicines10020425
Tuesday, February 8, 2022
In vivo overexpression of frataxin causes toxicity mediated by iron-sulfur cluster deficiency
Claudia Huichalaf, Tyler L. Perfitt, Anna Kuperman, Renea Gooch, Ramesh C. Kovi, Karrie A. Brenneman, Xian Chen, Dinesh Hirenallur-Shanthappa, Tiffany Ma, Basel T. Assaf, Ingrid Pardo, Tania Franks, Laura Monarski, Ting-Wen Cheng, Kevin Le, Chunyan Su, Suryanarayan Somanathan, Laurence O. Whiteley, Christine Bulawa, Marko J. Pregel, Alain Martelli; Molecular Therapy - Methods & Clinical Development,
2022, doi:10.1016/j.omtm.2022.02.002.

At the lowest tested dose, we observed moderate liver toxicity that was accompanied by progressive loss of transgene expression and liver regeneration. Together, our data provide insights into the toxicity of frataxin overexpression that should be considered in the development of a gene therapy approach for Friedreich’s ataxia.
Monday, February 7, 2022
Patients’ access to rare neuromuscular disease therapies varies across US private insurers
Nikoletta M. Margaretos, Komal Bawa, Natalie J. Engmann & James D. Chambers; Orphanet J Rare Dis 17, 36 (2022). doi:10.1186/s13023-022-02182-3
The high cost of providing coverage for rare disease therapies remains a key challenge for health insurers. This challenge will increase as regulatory agencies continue to approve increasingly large numbers of rare disease therapies and healthcare payers have to balance providing access with budget constraints.
The evaluated set of large US private insurers tended to apply coverage restrictions beyond the FDA label indication in their coverage policies for a set of rare NMD DMTs. Plans rarely applied the same criteria in their coverage policies for the same products. Inconsistent coverage criteria mean that patients with different insurers have variable access to the same therapies, which may have important consequences for patients who move from one plan to another.
Monday, January 31, 2022
Reata Pharmaceuticals Initiates Rolling Submission of New Drug Application with U.S. FDA for Omaveloxolone for the Treatment of Patients with Friedreich’s Ataxia
January 31, 2022. PLANO, Texas--(BUSINESS WIRE)-- Reata Pharmaceuticals, Inc. (Nasdaq: RETA), (“Reata,” the “Company,” “our,” “us,” or “we”), a clinical-stage biopharmaceutical company, today announced that the company has initiated a rolling submission of a New Drug Application (“NDA”) to the U.S. Food and Drug Administration (“FDA”) for omaveloxolone for the treatment of patients with Friedreich’s ataxia. The rolling submission allows Reata to submit portions of the regulatory application to the FDA for review on an ongoing basis. The company reiterates that it expects to complete the submission of the NDA by the end of the first quarter of 2022.
Plans to Complete Submission by the End of the First Quarter of 2022
If Approved, Omaveloxolone Would Become the First Therapy Indicated for the Treatment of Patients with Friedreich’s Ataxia
Sunday, January 30, 2022
Therapeutic Strategies Targeting Mitochondrial Calcium Signaling: A New Hope for Neurological Diseases?
Rodríguez LR, Lapeña-Luzón T, Benetó N, et al.; Antioxidants (Basel, Switzerland). 2022 Jan;11(1). DOI: 10.3390/antiox11010165. PMID: 35052668.
Calcium (Ca2+) is a versatile secondary messenger involved in the regulation of a plethora of different signaling pathways for cell maintenance. Specifically, intracellular Ca2+ homeostasis is mainly regulated by the endoplasmic reticulum and the mitochondria, whose Ca2+ exchange is mediated by appositions, termed endoplasmic reticulum–mitochondria-associated membranes (MAMs), formed by proteins resident in both compartments. These tethers are essential to manage the mitochondrial Ca2+ influx that regulates the mitochondrial function of bioenergetics, mitochondrial dynamics, cell death, and oxidative stress. However, alterations of these pathways lead to the development of multiple human diseases, including neurological disorders, such as amyotrophic lateral sclerosis, Friedreich’s ataxia, and Charcot–Marie–Tooth. A common hallmark in these disorders is mitochondrial dysfunction, associated with abnormal mitochondrial Ca2+ handling that contributes to neurodegeneration. In this work, we highlight the importance of Ca2+ signaling in mitochondria and how the mechanism of communication in MAMs is pivotal for mitochondrial maintenance and cell homeostasis. Lately, we outstand potential targets located in MAMs by addressing different therapeutic strategies focused on restoring mitochondrial Ca2+ uptake as an emergent approach for neurological diseases.
Friday, January 28, 2022
Comparing pipelines across ten rare neurological diseases
Pharmaceutical Technology. January 27, 2022.
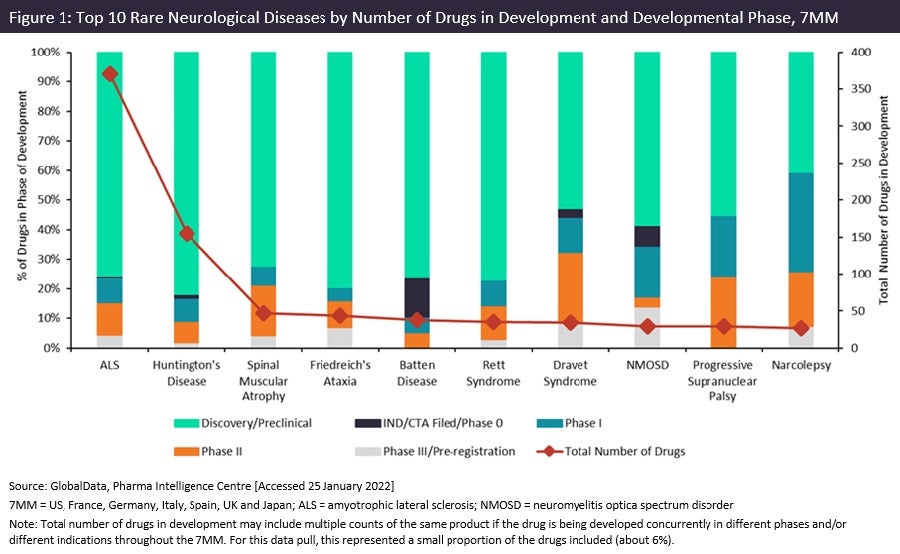
While much progress has been made in some rare neurological diseases, others are only beginning to see interest from developers.
Sunday, January 23, 2022
Mice harboring the FXN I151F pathological point mutation present decreased frataxin levels, a Friedreich ataxia-like phenotype, and mitochondrial alterations
Marta Medina-Carbonero, Arabela Sanz-Alcázar, Elena Britti, Fabien Delaspre, Elisa Cabiscol, Joaquim Ros & Jordi Tamarit. Cell. Mol. Life Sci. 79, 74 (2022). doi:10.1007/s00018-021-04100-5
We conclude that the primary pathological mechanism underlying the I151F mutation is frataxin deficiency, like in patients carrying GAA expansions. Therefore, patients carrying the I154F mutation would benefit from frataxin replacement therapies. Furthermore, our results also show that the FXNI151F mouse is an excellent tool for analyzing tissue-specific consequences of frataxin deficiency and for testing new therapies.
Friday, January 21, 2022
Hsa-miR223-3p circulating level is upregulated in Friedreich’s ataxia and inversely associated with HCLS1 associated protein X-1, HAX-1
Andrea Quatrana, Elena Morini, Francesca Tiano, Chiara Vancheri, Luca Panarello, Silvia Romano, Christian Marcotulli, Carlo Casali, Caterina Mariotti, Alessia Mongelli, Mario Fichera, Alessandra Rufini, Ivano Condò, Giuseppe Novelli, Roberto Testi, Francesca Amati, Florence Malisan; Human Molecular Genetics, 2022;, ddac005, doi:10.1093/hmg/ddac005
This study describes for the first time the association between hsa-miR223-3p and HAX-1 expression in FRDA, thus supporting a potential role of this microRNA as non-invasive epigenetic biomarker for FRDA.
Tuesday, January 18, 2022
Enfermedad cardiaca en la ataxia de Friedreich. Revisión diagnóstica y terapéutica
Sánchez Ortiz, M; Ibarra Reyes; López Pérez; Revista Ocronos. Vol. V. Nº 1–Enero 2022. Pág. Inicial: Vol. V; nº1: 91
Presentamos el caso de un varón de 27 años, con antecedentes de FRDA desde hace 11 años, válvula aorta bicúspide, hipertrofia ventricular izquierda y fumador. Su tratamiento farmacológico es citicolina de dispensación hospitalaria para la ataxia de Friedreich. Ingresado por deterioro de su clase funcional y palpitaciones de 15 días de evolución.
Monday, January 17, 2022
The attitude of patients with progressive ataxias towards clinical trials
Gilbert Thomas-Black, Andrada Dumitrascu, Hector Garcia-Moreno, Julie Vallortigara, Julie Greenfield, Barry Hunt, Susan Walther, Mackenzie Wells, David R. Lynch, Hugh Montgomery & Paola Giunti. Orphanet J Rare Dis 17, 1 (2022). doi:10.1186/s13023-021-02091-x
Knowledge of motivations for and barriers to trial participation as well as the acceptability of investigations, time commitments and routes of drug administration should inform better, more patient focused trial design. This in turn may improve recruitment and retention of participants to future trials.
Subscribe to:
Posts (Atom)

